Es un blog de Jorge Figueroa Apestegui Medico Cirujano General Especialista en Medicina Interna CMP:34170 RNE:031011Universidad Nacional Mayor de San Marcos 1990-2004
jorgeluisfigueroa1@outlook.com Hospital Arzobispo Loayza
Los meningiomas son tumores benignos de crecimiento lento, que representan el 15-20% de los tumores intracraneales. El 90% de los meningiomas son intracraneales. Es más común en mujeres en la sexta década de la vida.
La cirugía es el tratamiento de elección para la mayoría de los pacientes con meningiomas. La monitorización de la saturación venosa de oxígeno del golfo de la yugular (SjO2) forma parte de la monitorización multimodal en el paciente neurológico.
Introducción:
El meningioma es el tumor benigno más frecuente del sistema nervioso central en la práctica neuroquirúrgica. Es más común en mujeres en la sexta década de la vida. Los meningiomas comprenden el 15% de los tumores intracraneales primarios. Su incidencia general es de 2.3/100.000 habitantes para los meningiomas benignos y de 0.17/100.000 para los malignos (1), e incrementa con la radiación, presencia de neurofibromatosis tipo 2 y factores hormonales. El 90% de estos tumores se localizan en el compartimiento intracraneal supratentorial, fundamentalmente en las regiones parasagitales, la convexidad cerebral y las alas del esfenoides (2). Habitualmente son tumores sólidos, aunque pueden existir cambios quísticos en el 1.63 al 7.3%3.
La cirugía es el tratamiento de elección para la mayoría de los pacientes con meningiomas. Los objetivos de la cirugía son: la resección total del tumor, hueso y dura de alrededor implicados, y, si es posible, revertir o mejorar la clínica neurológica (4).
La monitorización de la saturación venosa de oxigeno del golfo de la yugular (SjO2) forma parte de la monitorización multimodal en el paciente neurológico.
Caso clínico
Presentamos el caso de una paciente de 61 años con antecedentes de hipertensión arterial en tratamiento con Ameride. A raíz de cuadro de alteración del habla y lenguaje se realizó RMN cerebral que informó de lesión extraaxial dependiente de hoz cerebral, con hipervascularización, sugestiva de hemangiopericitoma versus meningioma.
Se solicitó angioRMN con fase arterial y venosa para valorar arterias englobadas en el tumor además de la ocupación del seno longitudinal superior. La secuencia arterial observó una marcada hipertrofia de las arterias meníngeas medias de forma bilateral y simétrica, con desplazamiento caudal de las arterias pericallosas, y en la secuencia venosa no identificó flujo en la porción anterior del seno longitudinal superior. Se programó para exéresis de meningioma parasagital frontal izquierdo mediante craneotomía.
En quirófano se canalizó la arteria radial izquierda con la paciente despierta para monitorizar presión arterial invasiva (PAI) y la inducción anestésica transcurrió sin incidencias. La intubación orotraqueal con tubo orotraqueal nº 7.5 resultó fácil. Se canalizó vena yugular interna derecha sin incidencias. Dado que se trataba de un tumor muy vascularizado, se decidió monitorizar la saturación de la oxihemoglobina a nivel del bulbo de la vena yugular interna (SjO2) de forma continua mediante catéter en el golfo de la vena yugular interna izquierda.
Por la infraestructura de nuestro hospital no se comprobó radiológicamente su localización. Al realizar la craneotomía se observó gran vascularización, y al levantar el colgajo óseo se visualizó meningioma adosado a calota con lo que se abrió el seno en su trayecto sagital. Aunque hubo cierta dificultad en controlar la hemorragia se consiguió la exéresis completa del tumor. Durante la cirugía destacó gran labilidad hemodinámica y un sangrado importante que precisó la transfusión de 2 concentrados de hematíes. Además se registraron unos valores iniciales de saturación venosa de oxigeno del golfo de la yugular (SjO2) de 94-96% (normal: 55%-75%), que se mantuvieron altos durante toda la intervención y sólo descendieron hasta el 69% al final de la cirugía. La paciente se trasladó a Reanimación intubada y ventilada hasta realizar TAC craneal de control que evidenció cambios postquirúrgicos secundarios a craneotomía frontal, moderado neumoencéfalo frontal sin signos de efecto masa ni de sangrado intraparenquimatoso. La paciente fue extubada a las 24 horas sin incidencias.
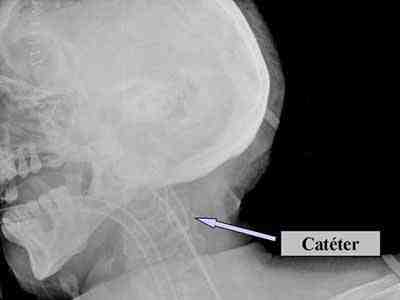
En reanimación permaneció con valores altos de saturación venosa de oxigeno del golfo de la yugular (SjO2), alrededor de 90% y la diferencia arterio- venosa del contenido de oxígeno (AVDO2) resultó inferior a 4 Vol% (valor normal 4-7 Vol%). En la radiografía de control se visualizó un bucle del catéter del golfo de la yugular con la punta situada en posición adecuada (figura 1). A pesar de su correcta localización, los valores de saturación venosa de oxigeno del golfo de la yugular (SjO2) permanecían elevados, y el cálculo de la AVDO2 < 4Vol% indicaba hiperemia., por lo que se mantuvo un estricto control de la presión arterial media y de la presión de perfusión cerebral.
Dado que la paciente permaneció asintomática y no se observó focalidad neurológica se decidió retirar el catéter a las 24 horas. La paciente fue dada de alta de Reanimación transcurridas 36 horas de la cirugía.
Discusión
Los síntomas causados por el meningioma se deben a la compresión del cerebro o la médula espinal. El tumor tiene predilección por ciertas regiones y produce síntomas y signos específicos según la localización del tumor.
En el 50-60% de los pacientes el diagnóstico puede sospecharse por cambios que se muestran en las radiografías de cráneo. La TAC es el método definitivo de diagnóstico que muestra el tumor como una masa homogénea de bordes bien definidos que realza con contraste. La resonancia magnética nuclear (RMN) con contraste permite identificar muy bien el meningioma y su relación con estructuras neurales y vasculares. La angiografía demuestra la vascularización del tumor y el suministro de sangre, aportando información para la futura cirugía y /o la posible embolización preoperatoria en ciertos casos. También permite determinar el grado de compromiso de la arteria carótida interna y sus ramas.
Las opciones de tratamiento incluyen la observación, la cirugía y la radiación:
Observación: Los pacientes de edad avanzada con tumores pequeños y asintomáticos pueden seguir controles por RMN anualmente. Dado que son tumores benignos y de crecimiento lento, no suelen causar ningún problema durante la vida de algunos pacientes.
– Cirugía: En pacientes más jóvenes, cuando los tumores están localizados en zonas críticas, y causan síntomas neurológicos (cefalea, alteraciones de la visión, trastornos convulsivos..),la extirpación quirúrgica es la mejor opción de tratamiento.
Radiación: Se aplica en pacientes que padecen tumores situados en regiones de difícil acceso o en pacientes que tienen un riesgo considerable para la cirugía. La radiocirugía estereotáctica también es un tratamiento a tener en cuenta.
Aunque los meningiomas son recurrentes, la resección completa incluyendo la eliminación del margen dural se asocia con una baja tasa de recurrencia.La neuromonitorización durante la cirugía incluye: PAI, presión venosa central (PVC) por vía yugular, subclavia o antecubital, bloqueo neuromuscular, end/tidal CO2, gasometrías (GSA) intermitentes, temperatura cerebral por termómetro timpánico o nasofaríngeo, diuresis mediante sondaje vesical, control del estado del flujo sanguíneo cerebral/oxigenación cerebral mediante saturación venosa de oxigeno del golfo de la yugular (SjO2), saturación regional de O2 (SRO2) y Doppler transcraneal (DTC).
La SjO2 proporciona información del metabolismo cerebral, ya que mide la relación entre el flujo sanguíneo cerebral (FSC) y el consumo metabólico cerebral de oxígeno (CMCO2). Es muy utilizada en los traumatismos craneales y se está introduciendo en los procesos neuroquirúrgicos. La vena yugular interna (VYI), se origina dentro del cráneo y se localiza en el cuello dentro de la vaina carotídea por detrás del esternocleidomastoideo y posterolateral a la carótida. La vena yugular interna recoge la mayor parte del flujo sanguíneo cerebral, con la salvedad de la sangre cerebral que drena hacia venas yugulares externas o venas vertebrales que representan un flujo mínimo.
El bulbo de la yugular es una dilatación en la base del cráneo y es el sitio de elección para obtener las muestras venosas. El 70% del flujo sanguíneo del bulbo de la yugular deriva del hemisferio cerebral ipsilateral y el 30% del contralateral, y sólo un 0.6-6% representa sangre extracerebral (venas faciales..). En la mayoría de los pacientes el drenaje derecho es el dominante.
La monitorización se realiza a través de un catéter de fibra óptica que mide la saturación de hemoglobina (Hb) en la sangre recogida en el bulbo de una de las venas yugulares internas, a través de espectrometría. El empleo de catéteres de fibra óptica permite obtener lecturas de forma continua o intermitente, sin embargo su principal inconveniente es que la correcta interpretación depende de la calibración y de la posición del catéter (5).
Estos catéteres consisten en un sistema de doble fibra óptica (uno emisor y otro receptor) junto con una vía lateral de infusión y extracción de muestras, con un calibre global de 4 French y una longitud de 40 cm. También se pueden utilizar catéteres venosos centrales habituales (sin fibra óptica) para mediciones de oximetrías discontinuas. El principio fundamental para la correcta interpretación de la saturación venosa de oxigeno del golfo de la yugular (SjO2) es que las muestras de sangre venosa tengan un origen exclusivamente cerebral, por lo que el catéter debe introducirse hasta alcanzar el bulbo de la yugular, lo cual minimiza el riesgo de contaminación por sangre extracerebral (6).
Los estudios efectuados a este respecto han demostrado que a nivel del bulbo de la yugular sólo un 2 a 3% de la sangre tiene un origen extracerebral. Cuando la punta se encuentra a más de 1 cm por debajo del bulbo, la contaminación con sangre extracerebral puede ser mayor a un 17%, lo que incrementa a un 50% cuando la punta se localiza por debajo de la quinta vértebra cervical. La localización de la punta del catéter obliga a tener controles radiológicos antes de obtener las muestras. En una radiografía cervical lateral, la punta del catéter debe localizarse al mismo nivel y por delante de la apófisis mastoides.
En caso de lesiones difusas debe colocarse el catéter en la vena yugular derecha, que recoge mayor flujo que la izquierda. En presencia de lesiones focales se debe colocar el catéter homolateral al hemisferio cerebral con mayor afectación. Otra opción es identificar mediante compresión yugular alterna el lado de drenaje predominante (el que produce mayor elevación de la presión intracraneal (PIC)) y colocarlo en esta yugular.
La saturación venosa de oxigeno del golfo de la yugular (SjO2) permite calcular otros parámetros: la oxigenación cerebral y el CMCO2, el FSC, el cociente FSC/CMCO2 y la diferencia arterio-venosa cerebral de O2 (AVDO2). La SjO2 es útil para detectar de forma precoz isquemia cerebral, valorar la capacidad de autorregulación del FSC y optimizar el tratamiento.
Los valores que se consideran normales de SjO2 oscilan entre 55%-75% con una AVDO2 de 4-7 ml O2 / 100 ml sangre. Los valores anormales de saturación venosa de oxigeno del golfo de la yugular (SjO2) se muestran en la Tabla 1:
Una disminución saturación venosa de oxigeno del golfo de la yugular (SjO2)< 55% indica que el aporte de oxígeno cerebral es insuficiente para la demanda metabólica, ya sea por una disminución del FSC (hipoxia cerebral) o por aumento del metabolismo cerebral: disminución de la presión de perfusión cerebral (PPC), anemia, hipoxia, hiperventilación, falta de sedación, fiebre, convulsiones.
Una saturación venosa de oxigeno del golfo de la yugular (SjO2)elevada > 75% indica que el FSC supera el consumo tisular (hiperemia cerebral), como sucede con el aumento de PPC, hiperoxia, infarto cerebral, hipotermia, muerte cerebral.
Tabla 1:
Saturación venosa de oxigeno del golfo de la yugular (SjO2) Normal (55-75%)
Interpretación: Flujo sanguíneo cerebral adaptado al consumo de oxígeno
Situación clínica: Compatible con la normalidad
Tratamiento: Mismo tratamiento
Saturación venosa de oxigeno del golfo de la yugular (SjO2)<55%
Interpretación: Disminución del FSC
Incremento CMCO2
Situación clínica: Hipoxia cerebral
Hiperactividad neuronal
Aumento del metabolismo cerebral
Tratamiento: Normoventilación
Tto. vasoespasmo, convulsiones, hipertermia, HTIC
Saturación venosa de oxigeno del golfo de la yugular (SjO2)>75%
Interpretación: Aumento FSC
Disminución CMCO2 Situación clínica: Hiperemia cerebral
Zonas de infarto
Muerte cerebral
Hipotermia
Compromiso en la liberación de O2
Error en la determinación (malposición del catéter, contaminación con sangre extracerebral, canulación de otro vaso venoso)
Tratamiento: Hiperventilar
Tto. HTIC
HTIC (Hipertensión intracraneal)
Por tanto, el valor de SJO2 es directamente proporcional al flujo sanguíneo cerebral e inversamente al consumo metabólico. Las técnicas de monitorización de la oxigenación cerebral como la SjO2 nos permiten iniciar y optimizar tratamientos en pacientes con grave compromiso del funcionalismo cerebral como TCE graves, tumores, hemorragias y otras lesiones ocupantes de espacio, que producen HTE, y así ajustar medidas terapéuticas como la hiperventilación, los agentes osmóticos y las aminas vasopresoras.
Las limitaciones de la monitorización de la SjO2 se muestran en la Tabla 2.
Las complicaciones asociadas a la utilización de las técnicas de oximetría yugular son extremadamente bajas (7). Entre ellas las propias de la técnica de canalización como: punción de la arteria carótida, hematoma arterial o venoso, lesión del nervio frénico o laríngeo recurrente, Síndrome de Horner, enrollamiento del catéter, punción de vena errónea (vena facial o seno venoso transversos); infección del catéter; y trombosis vascular
Limitación: Mezcla incompleta
Etiología: La muestra de sangre venosa puede no representar todo el cerebro si hay drenaje venoso asimétrico
Manejo: Canular la vena yugular interna más importante (derecha) o en el lado más dañado
Limitación: Contaminación extracerebral
Etiología: 3% de la sangre yugular está contaminada con sangre extracerebral
Manejo: Confirmación radiológica
Limitación: Efecto Bohr
Etiología: Falsa elevación de la SjO2 por desviación a la izquierda de la curva de disociación de la hemoglobina
Manejo: Se detecta midiendo presión del bulbo de la yugular demasiado baja (<27 mmHg)
Limitación: Medición global
Etiología: Si lesión cerebral focal, SjO2 puede no dar información de la región lesionada
Manejo: Medición del lactato A-V puede ayudar como indicador de metabolismo anaerobio
Limitación: Insensible a flujo infratentorial
Etiología: El tallo cerebral y cerebelo contribuyen un poco al flujo de salida de sangre del cerebro
Manejo: Es un valor limitado para monitorizar pacientes con lesiones de tallo cerebral
Limitación: Errores de monitorización
Etiología: El catéter puede estar contra la pared de la vena enrollado en sí mismo o con fibrina en la punta
Manejo: Se puede necesitar reposicionar el catéter, recalibrar o reheparinizar el catéter.
Tabla 2. Limitaciones de la SjO2
En el caso que presentamos, la presencia del bucle en el catéter, no afectó la posición del extremo del mismo, evidenciado por la radiografía de columna cervical. Los valores de SjO2 superiores a 90% (valores muy “arteriolizados”) observados en Reanimación, se debieron a hiperemia, confirmada con el cálculo de la AVDO2.
Actualmente, la neuromonitorización ha demostrado ser útil y segura, mejorando la morbimortalidad, debido a que se detecta la isquemia cerebral de forma temprana. Sin embargo, uno de los principales problemas y errores durante la monitorización de la SjO2 es que la punta del catéter no esté colocada en el bulbo de la yugular. Este hecho nos puede llevar a falsos diagnósticos y, en consecuencia, a tratamientos erróneos. Por eso creemos muy recomendable no iniciar ningún tratamiento hasta que se haya comprobado con una prueba de imagen que la punta del catéter está correctamente colocada.
Figura 1. Visualización por Radiografía de cráneo del bucle del catéter del golfo de la yugular.
La incidencia de tromboembolismo pulmonar (TEP) en países desarrollados se ha estimado en 1 caso por cada 1.000 habitantes y año .Esta incidencia aumenta al 1% en ancianos. (1,2,3)
La mortalidad a los 3 meses, según el International Cooperative Pulmonary Embolism Registry fue del 17,5%. (2) El 75-90% de los fallecimientos tienen lugar en las primeras horas de producirse el tromboembolismo pulmonar (TEP), por ello la mortalidad del tromboembolismo pulmonar (TEP) sigue siendo importante y consecuentemente el TEP debe ser precozmente diagnosticado y tratado adecuadamente.
DOLOR TORÁCICO CON CORONARIAS SANAS. SITUACIONES CLÍNICAS
El diagnóstico es difícil, ya que puede acompañar o simular otras enfermedades cardiopulmonares pero se ha de realizar un diagnóstico precoz del tromboembolismo pulmonar (TEP), para lo cual la sospecha clínica sigue siendo fundamental .Las pruebas diagnósticas no invasivas tienen una baja sensibilidad y especificidad, pero nuevas alternativas como la determinación de dímero D o la TAC helicoidal incrementan la precisión diagnóstica. (3,4)
DESCRIPCIÓN DEL CASO
Reportamos el caso de un varón de 64 años que ingresa desde vía pública mediante código IAM y que como antecedentes de interés presenta diabetes mellitus II , dislipemia y exfumador
Su historia cardiológica se resume en angina de esfuerzo de 2 años de evolución por la que se hizo una ergometría siendo negativa. Hace 2 meses en el preoperatorio para ser intervenido de estenosis uretral secundaria a hipertrofia benigna de próstata se detecta una alteración electrocardiográfica por lo que se realiza ergometría y cateterismo siendo ambos negativos también.
Actualmente estando bien previamente, presenta estado pre sincopal que al recuperarse el paciente explica un dolor opresivo centrotorácico y disnea.
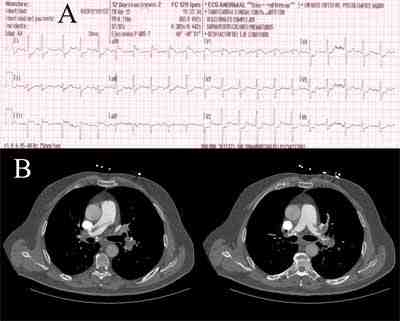
Se avisa al SEM que le realiza un electrocardiograma (ECG) con dolor en la ambulancia donde se describe un ritmo sinusal, eje izquierdo, 100 latidos por minuto (lpm), elevación ST D3 y AVR, Q en D3 y descenso difuso ST en el resto de derivaciones (FIGURA A) y se inicia doble antiagregación y morfina endovenosa mejorando la clínica, no se inicia solinitrina por episodio de hipotensión, y a la exploración física muestra una presión arterial de 130/89 mmHg, 95 latidos por minuto, una temperatura de 36,5ºC y una saturación de oxígeno del 96%.
Por ello ingresa directamente a la sala de hemodinámica donde se hace el cateterismo cardíaco no evidenciando lesiones significativas. En el ECG post cateterismo se evidencia una retrogradación de los cambios del ST y mejoría clínica.
La Rx de tórax portátil es de mala calidad y sin alteraciones valorables, la analítica y gasometría con mascarilla de alto flujo evidencia una acidosis metabólica (ph 7.27, pCO2 38, pO2 145, HCO3 18, EB -8)y una alteración difusa de la función hepatobiliar
Se le realiza una ecografía transtorácica visualizando una dilatación del ventrículo derecho, ventrículo izquierdo de pequeño tamaño sugiriendo hipovolemia, dilatación de la vena cava inferior (2.1cm) sin colapso respiratorio. Se sospecha de tromboembolismo pulmonar, por ello se realiza también Tac torácico donde se confirma tromboembolismo pulmonar (TEP) masivo bilateral con trombo acabalgado entre la arteria pulmonar derecha e izquierda .FIGURA B
Fue ingresado y recibió tratamiento con heparina de bajo peso molecular endovenosa, produciéndose una buena evolución y recuperación del paciente.
Figura A: ECG de inicio. Figura B: Tac torácico
Tromboembolismo pulmonar. TEP. TAC. ECG
DISCUSIÓN
Este caso destaca por la gravedad del cuadro, ya que en su inicio se pensó en un síndrome coronario agudo por afectación del tronco común debido a los cambios electrocardiógrafos anteriormente descritos y por ello se activó el código IAM para realizar reperfusión inmediata, observándose que tras realizar el cateterismo con administración de heparina sódica endovenosa se resolvió el cuadro electrocardiográfico y clínico. Además resaltar que la ecografía fue clave para la sospecha del tromboembolismo pulmonar (TEP) y así posteriormente poderlo confirmar con un Tac helicoidal.
El tromboembolismo pulmonar (TEP) sigue siendo un desafío diagnóstico y terapéutico por ello recordar que la sospecha de TEP es clave para el diagnóstico en pacientes con factores de riesgo y clínica sugerente.
El Síndrome de Charles Bonnet se caracteriza por alucinaciones visuales, sin trastornos cognitivos ni psiquiátricos acompañantes. Suele tratarse de pacientes ancianos con severas alteraciones visuales (1,2,3,4,5,6), que permanecen ingresados en hospitales, en ambientes con poca luz y apartados de su ambiente familiar y social habitual. El objetivo de este trabajo es describir tres casos de pacientes afectos de este síndrome en nuestras unidades de cuidados intensivos.
INTRODUCCIÓN
En 1759, Charles Bonnet, describió por primera vez el caso de un anciano parcialmente ciego, sin deterioro orgánico cerebral apreciable y con alucinaciones visuales, que reconocía como irreales. Posteriormente se han descrito en la literatura médica casos similares englobándolos bajo la denominación de síndrome de Charles Bonnet (1,2,3,4,5,6) . Sin embargo, este síndrome es infrecuentemente reconocido o mal diagnosticado, agravándose este hecho en las unidades de cuidados intensivos. En este ambiente cuando un paciente refiere tener alucinaciones visuales se piensa de inmediato en un delirio, una psicosis con o sin demencia o una intoxicación medicamentosa.
El objetivo de este artículo consiste en describir tres casos de síndrome de Charles Bonnet detectados en nuestras unidades de cuidados intensivos
CASO 1
Mujer de 74 años, con antecedentes personales de hipertensión arterial, diabetes insulinodependiente y retinopatía diabética con repercusión importante sobre su visión, no refiere alergias medicamentosas ni hábitos tóxicos. Ingresa en UCI por dolor centrotorácico, elevación de las troponinas hasta 2,5ng/ml e infradesnivelación del ST en DII, DIII y aVF. En tratamiento con nitroglicerina, heparina intravenosa, aspirina, plavix y tirofiban, comienza a las pocas horas de su estancia en UCI a referir alucinaciones visuales. Describía ver figuras geométricas de colores vivos que iban cambiando de forma y localización, produciéndole este fenómeno una gran ansiedad, aunque las reconocía como irreales y que desaparecían cuando cerraba los ojos.
La exploración física era compatible con la normalidad, sin signos de bajo gasto cardíaco ni insuficiencia cardíaca. Analíticamente solo era de reseñar la elevación en la curva de la troponina I y glucemia de 170mg/dl. No tóxicos en orina Exploración neurológica, cognitiva y psiquiátrica compatibles con la normalidad.
Tomografía axial computerizada cerebral normal al igual que la resonancia nuclear magnética. Reconociendo la propia paciente la irrealidad de las alucinaciones se la tranquilizó y se diagnosticó de síndrome de Charles Bonnet, desapareciendo de forma progresiva las mismas en las siguientes horas y sin tener que administrar neurolépticos o sedantes, evolucionó favorablemente y fue dada de alta al tercer día de estancia en UCI tras realización de cateterismo y colocación de stent en coronaria derecha. Cuando iba a ser dada de alta reconoció haber tenido episodios similares en casa, aunque nunca lo comentó ni consultó con nadie porque pensaba que sus hijos pensarían que estaba demenciándose.
CASO 2
Varón de 72 años de edad, sin hábitos tóxicos, hipertenso y con pérdida importante de la visión desde hace dos años por degeneración macular senil. Ingresa en UCI tras caída fortuita por las escaleras, seguida de náuseas, vómitos, mareos, cefaleas occipitales y cuadro de visión de escenas alucinatorias. Describía ver a su padre corriendo por una pradera muy verde, animales en su habitación y círculos de colores que no cesaban de girar. El paciente reconocía la irrealidad de las visiones.
La exploración neurológica era compatible con la normalidad. Analítica normal. Ausencia de tóxicos en orina. La tomografía axial computerizada evidenció pequeño hematoma occipito-parietal derecho. Se instauró tratamiento con gabapentinas y al segundo día de estancia en UCI desapareció la clínica, siendo dado de alta el paciente al tercer día de su ingreso sin déficit neurológico alguno
CASO 3
Mujer de 69 años de edad, con antecedentes de hipertensión, diabetes insulinodependiente, dislipemia familiar, pérdida de visión por retinopatía de etiología proliferativa, cardiopatía valvular consistente en estenosis mitral severa, que ingresa en UCI para control postoperatorio inmediato de resección de adenocarcinoma de colón, realizándose anastomosis latero-lateral sin incidencias reseñables durante el acto quirúrgico.
A las pocas horas de estar en UCI comienza a referir que ve animales en su habitación y personas que están paseando de un lado a otro. Reconoce que estas visiones son irreales, pero le producen ansiedad. Refiere haber tenido hace dos años un cuadro similar que duró tres días, pero que no consultó ni se lo dijo a nadie.
Es vista por neurólogo y psiquiatra que no evidencian patología. Analíticamente no hay alteraciones. No tóxicos en orina. Tomografía axial computeriza y resonancia nuclear magnética normales. Se tranquiliza a la paciente, se le diagnostica de síndrome de Charles Bonnet y tras ser informada la propia paciente se niega a tomar medicación para este cuadro. Las alucinaciones desaparecen al ser dada de alta de UCI
COMENTARIOS
La prevalencia del síndrome de Charles Bonnet es del 11-13% en personas con pérdidas visuales parciales y del 1 al 2% en la población geriátrica (1,2,5). Muchos de estos pacientes, presentan alucinaciones visuales en su casa,durante periodos variables de tiempo o en distintos momentos a lo largo de su evolución, pero prefieren ocultar el trastorno temiendo ser tildados de enfermos psiquiátricos, como ocurrió en nuestros casos 1 y 2.Los criterios tradicionales para su diagnóstico exigían que la vía y la corteza visual estuvieran indemnes (1,2). Sin embargo se han descrito casos de enfermos con hemorragias o infartos 6 u otras alteraciones en la corteza cerebral occipital, como sucede en el caso 2.
El compromiso de la visión en el síndrome de Charles Bonnet es severo pero no total, los pacientes conservan la visión luz y sombra o cuentan dedos. Se describen casos en la literatura médica, en que el paciente deja de tener alucinaciones una vez queda totalmente ciego o una vez recupera o mejora la visibilidad (4,5) .Todos nuestros pacientes tenían severa afectación de la visibilidad. Las patologías oculares más frecuentes asociadas a este síndrome son la degeneración macular senil, la retinopatía diabética, el glaucoma y las afectaciones corneales, circunstancias que se da en nuestra casuística.Las alucinaciones que se describen en la literatura médica, al igual que en los nuestros, suelen ser complejas, variables y coloridas, a veces relacionadas con figuras geométricas, con escenas familiares, con paisajes o con animales (1,2,3,4,5,6). Pueden ser agradables o desagradables. En ocasiones, desaparecen cuando el paciente cierra los ojos o cuando dirige la mirada hacia otro lado, como sucede en nuestro primer caso. Siendo premisa indispensable para el diagnóstico de este síndrome, que el paciente sea consciente de que lo que ve es irreal. Aunque en la mayoría de los casos suele crear gran ansiedad
La frecuencia de las alucinaciones varía entre dos episodios al año o varios por día durando desde pocos minutos a varias horas. Siendo más frecuente hacia el anochecer, cuando hay poca luz y durante la inactividad y la soledad, condiciones que hacen idónea la aparición de estos cuadros durante internamientos hospitalarios y en las unidades de cuidados intensivos. En estos servicios por las características estructurales de las unidades, clínicas y terapéuticas de los pacientes, ese síndrome es frecuente que pase inadvertido o se confunda con cuadros de delirium, psicóticos o iatrogénicos medicamentosos.
El diagnóstico diferencial del Síndrome de Charles Bonnet debe realizarse con alucinaciones por fármacos, epilepsia, migrañas, accidentes cerebro vasculares, demencias degenerativas, delirios o cuadros psicóticos.
La forma de tratar a estos pacientes es tranquilizarlos y explicarles que es lo que sucede para que disminuya su grado de ansiedad y sus temores. En ocasiones los neurolépticos y los antiepilépticos han dado buenos resultados (6).
En las unidades de cuidados intensivos es importante sospechar la posibilidad de esta etiología en pacientes que refieran alucinaciones visuales y tengan deterioros visuales, porque simplemente tranquilizándolos se puede evitar recurrir a sedaciones innecesarias cuando no peligrosas, dadas las características y edad de esta población. Además las unidades de cuidados intensivos y probablemente cualquier internamiento de este tipo de pacientes sean los lugares propicios para la aparición de estos cuadros dada sus características de escasa luminosidad, aislamiento de la persona…etc.
En cuanto a la fisiopatología de las alucinaciones podría ser un fenómeno de desaferentización, producido por incremento de la excitabilidad de las neuronas desaferentizadas, ocasionada por cambios moleculares como puede ser el aumento del número de receptores de la membrana postsináptica ,o, bioquímicas como puede ser el incremento del N-Metil-D aspartato y la disminución del acido gamma-aminobutírico (1,2,3,4,5,6).
Si bien el diagnóstico de este síndrome no es sencillo, porque no existen criterios clínicos establecidos y en segundo lugar porque los episodios alucinatorios son variables en duración, frecuencia y características, incluso en el mismo paciente; pretendemos recordar su existencia y señalar de forma clara que no todos los pacientes con alucinaciones visuales en UCI tienen delirios o cuadros psicóticos y necesitan ser sedados; circunstancias éstas que pueden ser reproducibles en geriátricos e internamientos hospitalarios de pacientes con pérdida importante de visión y edades avanzadas.
Torsades de pointes por QT largo y apoplejía hipofisaria
El síndrome de QT largo se caracteriza por una prolongación de este intervalo en el electrocardiograma, definido como un QTc>450 ms en varones y >470 ms en mujeres, que predispone al desarrollo de arritmias ventriculares del tipo torsades de pointes (1-4). Se diferencian dos grandes grupos etiológicos: El QT largo congénito asociado a mutaciones genéticas y, la variante adquirida asociada sobre todo a la toma de fármacos (3), alteraciones electrolíticas (1,2,4) (hipopotasemia, hipomagnesemia e hipocalcemia), trastornos neurológicos (1,2,4), cardiológicos (1,2,3) y más raramente endocrinos (5-7). Presentamos el caso de un varón de 62 años, que acude al servicio de urgencias por bajo nivel de conciencia y fiebre. Su familia refirió dos episodios de pérdida de conciencia y pico febril de 38,5ºC, precedido de vómitos. Sus antecedentes personales reseñables eran fibrilación auricular e hipertensión arterial. A la exploración física se observó a un paciente sin focalidad neurológica, bradipsíquico, con lenguaje coherente, piel seca, tensión arterial 70/35mmHg, frecuencia cardíaca de 54lpm y afebril (en ese momento), resto de la exploración anodina.
Estando monitorizado en el Servicio de Urgencias se observa torsades de pointes y posterior fibrilación ventricular, por lo que se procedió a desfibrilar e iniciar perfusión de sulfato de magnesio, tras lo cual ingresa en UCI, donde presenta dos episodios más de torsades de pointes. Se le realiza ECG evidenciándose ACxFA a 50lpm con QTc de 630ms y, una analítica en la que destaca: sodio 116mmol/l, calcio corregido 8,5 mg/dl, magnesio 1,5 mg/dl.
La TAC evidenció apoplejía hipofisaria secundaria a sangrado de posible macroadenoma. Se solicitó estudio hormonal que objetivó panpituitarismo: TSH 0,13uU/ml (0,27-4,2), T3 libre 0,5pg/ml (1,8-4,6), FSH 0,51mUI/ml (1,37-13,58). LH 0,15mUI/ml (1,8-8,2), cortisol basal 2ug/dl (6,2-19,4). Ante estos resultados se inició tratamiento con levotiroxina 100ug/día e hidrocortisona 50 mg/ 8 horas, que corrigió el QT.
Se consultó el caso con neurocirugía que optó por tratamiento con radioterapia del macroadenoma hipofisaria. Y dado que está documentado el alto riesgo de muerte súbita por arritmias ventriculares, se decidió implantar desfibrilador por el riesgo de recidiva de la torsade de pointes ante la situación de estrés a la que iba a estar sometido este paciente, durante y después del tratamiento radioterápico. A los dos meses del alta hospitalaria, el paciente acudió a revisión de control del desfibrilador, objetivándose ACxFA con QTc de 410ms, estaba asintomático y no había presentado arritmias ventriculares.
La apoplejía hipofisaria es rara, cuando aparece puede ser isquémica o hemorrágica, siendo, en estos casos, la mayor parte de las veces secundaria a sangrado de macroadenoma, cursa con panhipopituitarismo. La clínica suele ser insidiosa y en la literatura no hemos encontrado ningún caso asociado a Torsades de Pointes, si bien si que han sido descritas en casos de silla turca vacía (4,5) y otras patologías asociadas a alteraciones endocrinológicas (4,5,6), como el hipotiroidismo y las patologías del eje corticoideo-suprarrenal (6,7). Por su importante interacción hormonal para la respuesta sistémica y cardiovascular ante el estrés, es importante el tratamiento de sustitución hormonal tiroideo y corticoideo (4-7) lo más precozmente posible.
Presentamos un caso de panhipopituitarismo secundario a apoplejía hipofisaria que ingresó con clínica de insuficiencia suprarrenal aguda (shock hipovolémico, bajo nivel de conciencia, fiebre y alteraciones hidroelectrolíticas) que desarrolló torsades de pointes por QT largo adquirido, secundario a déficit hormonal del eje cortico-tiroideo, con corrección del QT tras tratamiento hormonal sustitutivo, lo que consideramos nos debe tener en alerta, ante cualquier paciente hipotiroideo o con insuficiencia suprarrenal que ingrese en los servicios de medicina intensiva y que presente un QTc largo, por su riesgo elevado de presentar torsades de pointes.
El síndrome de Percheron o infarto talámico bilateral sincrónico se considera infrecuente y de difícil diagnóstico clínico. Presentamos el caso de una paciente de 71 años con disminución del nivel de conciencia con respuesta al estimulo verbal e hipersomnia intensa al despertar el día del ingreso. Tras las pruebas complementarias se observan lesiones isquémicas agudas en ambos tálamos, compatibles con obstrucción del la arteria de Percheron.
Autores: Rosa Ana Serrano Benavente. Médico de familia adjunto a la UGC de Cuidados Críticos y Urgencias del Hospital Comarcal de la Axarquía.
INTRODUCCIÓN
Los tálamos se ubican a ambos lados de la línea media formando las paredes laterales del tercer ventrículo y se extienden lateralmente hasta la cápsula interna. La lámina medular interna, lo cruza en sentido anteroposterior y lo divide en cuatro grupos de núcleos: anteriores, laterales, mediales e intralaminares. Además, se distinguen otros 2 núcleos: el reticular y los de la línea media. El grupo anterior y el medial, participan en el aprendizaje, las funciones mnésicas y el control emocional.
El lateral, en la sensibilidad y el movimiento, mientras que el núcleo intralaminar y el de la línea media presentan proyecciones difusas a la corteza y ganglios basales, y regulan la actividad cortical. La irrigación del tálamo es de tipo terminal y está dada por cuatro arterias. La arteria tálamo perforante o paramediana, que se origina en la primera porción de la arteria cerebral posterior (ACP), irriga la región medial del tálamo. La arteria tubero-talámica o polar, rama de la comunicante posterior, irriga la porción anterior y en un 30-40% de la población no existe, siendo reemplazada por la arteria paramediana.
La región posterolateral, es irrigada por la arteria tálamo-geniculada, y el territorio paradorsal, por la arteria coroidea posterior; ambas ramas de la arteria cerebral posterior (ACP). Lesiones isquémicas talámicas bilaterales pueden explicarse por la oclusión de una variante anatómica de las arterias paramedianas. Estas arterias nacen hasta en un 45% de los casos de una misma arteria cerebral posterior (ACP), ya sea en forma independiente o a partir de un tronco común. Esta última variante es conocida como arteria de Percherón, y su oclusión origina infartos talámicos mediales bilaterales, relativamente simétricos
Presentamos el caso de una paciente con oclusión de la arteria de Percherón, tras el cual desarrolló deterioro cognitivo.
CASO CLÍNICO
Anamnesis
Mujer de 71 años, con antecedentes personales de: No alergias medicamentosas conocidas. No hipertensión arterial. No diabetes mellitus. No dislipemia. No enfermedades cardiacas ni pulmonares. Osteoporosis. Migraña. Independiente para todas las actividades de la vida diaria. No intervenciones quirúrgicas. En tratamiento con: calcio-vitamina D, paracetamol. No hábitos tóxicos.
La paciente acude al servicio de urgencias de nuestro hospital trasladada en ambulancia por episodio de disminución del nivel de conciencia al intentar despertarla por la mañana. El marido refiere que el día anterior no presentaba ninguna sintomatología y por la noche comenzó con hipersomnia importante con cefalea leve asociada, lo que motivo que le ayudaran a meterse en la cama a dormir. Esta mañana al ir a despertarla la paciente presentaba disminución del nivel de conciencia con respuesta a la estimulación verbal intensa con tendencia al sueño posterior. No observan alteración de motora y reconocen lenguaje algo enlentecido. No fiebre. No síntomas respiratorios, ni digestivos ni urinarios asociados los días previos. No traumatismos. No ingesta de tóxicos ni medicación conocido.
Exploración física
TA: 119/70, FC: 60 latidos por minuto (lpm), frecuencia respiratoria: 17 respiraciones por minuto (rpm), SAT 02: 98%, Temperatura: 36,5ºC
REG; estuporosa, apertura ocular a estimulo verbal intenso con lenguaje enlentecido. BH y P. Eupneica. Normocoloreada. No exantemas ni petequias.
C-C: faringe normocolórica, no adenopatías, Pulsos carotídeos simétricos palpables sin soplos. No ingurgitación yugular. No bocio.
Tórax: Tonos cardíacos puros y rítmicos a 60 latidos por minuto (lpm), murmullo vesicular conservado sin ruidos patológicos.
Abdomen: blando y depresible, no masas ni megalias, no doloroso, puñopercusión renal negativa, pulsos femorales simétricos palpables
Extremidades: no edemas, pulsos periféricos palpables, no signos de trombosis venosa profunda (TVP)
Exploración neurológica: Estado mental: obnubilada, desorientada en tiempo espacio y persona. Lenguaje enlentecido responde parcialmente con monosílabos. Pupilas isocóricas normorreactivas, permanece con ojos cerrados, apertura ocular con estimulo verbal intenso. Pares craneales aparentemente normales. Sistema motor: Fuerza, sensibilidad tono y trofismo muscular normales. Marcha y Romberg no valorado por imposibilidad de bipedestación de la paciente; Reflejos osteomusculares normales y cutaneoplantar flexor. No signos meníngeos.
Pruebas complementarias
ECG: Ritmo sinusal a 60 latidos por minuto (lpm), eje normal, sin alteración de la repolarización.
Analítica: Bioquímica: Glucosa 85 mg/dl, Creatinina 0,56 mg/dl, Sodio 138 mEq/L, Potasio 5,2 mEq/L, Calcio 9,1 mg/dl, magnesio 1,94, TSH 1,2, CK 60,0 u/L Hemograma: Leucos 4680 (60%N, 29%L) Hemoglobina 13,7g/dl, Hematocrito 42%, VCM 91, HCM 27,8, Plaquetas 301000. Estudio de coagulación: Tiempo de protrombina 12,1 segundos, Actividad de protrombina 91,9%, INR 1,1, TPTA 26,7 segundos, TPTA ratio 0,9.
Rx tórax: índice cardiotorácico normal, no derrame pleural, no condensaciones ni infiltrados.
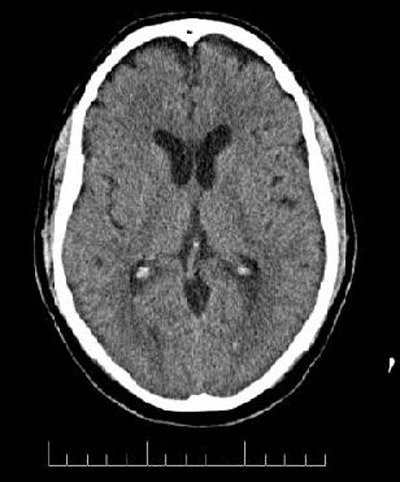
Tac craneal: se han realizado cortes contiguos de 2,5mm de grosor a nivel de fosa posterior, y de 7,5mm hasta el vértex sin contraste intravenoso. Signos de atrofia cortico-subcortical moderada. Lesión hipodensa por delante del asta anterior del Ventrículo lateral derecho de probable origen vasculo-isquémico crónico. No se aprecian otras lesiones focales intracraneales. Fosa posterior sin alteraciones. Sin otros hallazgos radiológicos a destacar en el momento actual. Conclusión: Cambios crónicos a correlacionar con antecedentes y factores de riesgo vascular (fig. 1)
Punción lumbar: hematíes 110, leucos 0, glucosa 54, proteínas 20, no se observan gérmenes.
RMN craneal: microinfartos solitarios en sustancia blanca profunda, así como lesiones isquémicas agudas de localización paramedial en ambos tálamos y mesencéfalo anterior, compatibles con obstrucción de la arteria de Percherón (fig. 2)
Diagnóstico diferencial
Ante los antecedentes de la paciente, la clínica y exploración física y la normalidad de las pruebas realizadas se nos planteó como primera opción diagnóstica Enfermedad cerebrovascular isquémica; sin poder descartar o contemplar otras opciones diagnósticas como:
-Etiología metabólica: encefalopatía de Wernicke, Creutzfeldt-Jakob, Mielinolisis osmótica, Enfermedad de Wilson, Enfermedad de Leigh, MELAS.
Juicio clínico
Síndrome de Percherón o infarto paramedial talámico bilateral.
Evolución
La paciente permaneció en observación durante 24 horas con mejoría del nivel de conciencia, amnesia de lo ocurrido e hipersomnia. Ante la sospecha del origen isquémico de la patología se inició tratamiento antiagregante con AAS 100mg. Tras valoración por Servicio de M Interna se decide ingreso en planta.
Durante el ingreso la paciente presenta buena evolución persistiendo deterioro cognitivo leve con desorientación temporal, es capaz de invertir series con lentitud, sin lograr retener nueva información, pero su lenguaje es fluido y sin parafasias. En el seguimiento posterior de la paciente (hasta la fecha), no ha habido progresión del deterioro cognitivo y no han aparecido otros nuevos hallazgos en el examen neurológico. El control con imágenes no ha demostrado aumento de la atrofia cortico-subcortical ni nuevas lesiones isquémicas. Puede desempeñar normalmente las actividades de la vida diaria y no acusa otras molestias.
DISCUSIÓN
El tálamo contiene núcleos estratégicos que integran múltiples funciones corticales importantes. Los infartos isquémicos bilaterales del tálamo son infrecuentes, siendo la causa más probable la oclusión de la arteria de Percherón. Ésta es una variante anatómica que permite la irrigación bilateral de los tálamos, a partir de un tronco común de origen asimétrico en la arteria cerebral posterior. Estas lesiones dan origen a una variedad de manifestaciones clínicas entre las cuales se ha descrito la demencia por infarto estratégico. Es una oclusión de la variante anatómica de las arterias paramedianas o de Percherón ocasionando infartos talámicos mediales bilaterales, relativamente simétricos. Este tipo de infarto representa el 0,6% de todos los primeros episodios de accidente vascular cerebral.
Su principal causa es la oclusión de la arteria de Percherón causada por la enfermedad de pequeño vasos asociadas a factores de riesgo cardiovascular o bien por cardioembolismo. La clínica es variable, dependiendo del territorio vascular afectado. Durante la fase aguda la sintomatología más frecuente es hipersomnia, parálisis ocular, ataxia moderada de la marcha, déficit de atención, aprendizaje y memoria. La presentación de los trastornos neurocognitivos se evidencian una vez resuelto el compromiso de conciencia inicial. Las alteraciones neurológicas suelen revertir paulatinamente, sin embargo los trastornos ejecutivos y la amnesia puede ser severa y persistente dando origen a una demencia vascular.
Presentamos el caso de una paciente con infarto talámico bilateral, tras el cual desarrolló leves alteraciones neurocognitivas con posible progresión a una demencia vascular a corto plazo. Un infarto talámico paramediano bilateral sincrónico debe incluirse siempre en el diagnóstico diferencial de enfermos con bajo nivel de consciencia y TC craneal sin lesiones agudas objetivables, especialmente cuando se han descartado causas sistémicas, y ante la presencia de hallazgos clave, como los trastornos oculomotores o signos focales de cualquier área.
CONCLUSIÓN
La comunicación de este caso refuerza la necesidad de tener siempre en cuenta la posibilidad en pacientes con disminución del nivel de conciencia. Cuando tiene lugar un cuadro isquémico dependiente de la arteria de Percheron las manifestaciones neurológicas que suelen aparecer son la afectación del nivel de conciencia con cambios fluctuantes en el mismo, incluyendo el coma, y además suele existir afectación del lenguaje, así como alteración del estado de ánimo en forma de apatía, y otras manifestaciones como afectación de pares craneales oculomotores, trastornos del movimiento (disquinesias), afectación de la memoria (amnesia) y del sueño (hipersomnia).
Esta forma de presentación es muy aproximada a la del caso descrito y confirma la necesidad de conocer el territorio irrigado por una arteria como la de Percheron, puesto que las lesiones que desencadena suelen ser alteraciones talámicas bilaterales. Reconocer esta afectación bilateral en los estudios de RM es fundamental para caracterizar el cuadro; además, cabe destacar que existen pocos casos descritos en la literatura de hiperintensidad bilateral en tálamo y la escasa utilidad que han demostrado otras pruebas de neuroimagen.
Feocromocitoma: diagnóstico y tratamiento en un paciente ingresado en la Unidad de Cuidados Intensivos
Objetivo: presentar una patología infrecuente que, en nuestro caso, tiene un debut grave.
Material y métodos: paciente de 45 años de edad que ingresa en la Unidad de Cuidados Intensivos por un síndrome coronario agudo con elevación del segmento ST. Tras tratamiento médico y de reperfusión coronaria persisten cifras tensionales muy elevadas (210/120 mmHg) que no se controlan con tratamiento médico. Se inicia estudio de hipertensión arterial, con el hallazgo de masa suprarrenal derecha.
Caso clínico:
Anamnesis:
Paciente de 46 años sin antecedentes personales de interés, acude al Servicio de Urgencias por referir desde hace 2-3 días episodios ocasionales de dolor centrotorácico opresivo de 1-2 minutos de duración, no irradiado, acompañado de sudoración fría, palidez y vómitos. El dolor disminuye pero sin desaparecer por completo, por lo que acude al servicio de Urgencias.
A su llegada presenta dolor torácico. En la exploración física destaca, sudoración, palidez cutánea, hipertensión arterial (160/100 mmHg) y fiebre de 38ºC, que cede con paracetamol 1 gramo intravenoso. En pruebas complementarias destaca, en electrocardiograma imagen de bloqueo de rama derecha, descenso del segmento ST de 2 mm en las derivaciones de V2 a V5.
Con estos hallazgos ingresa en UCI.
Exploración al ingreso:
Tensión arterial 210/120 mmHg, frecuencia cardíaca 150 latidos por minuto, saturación de oxígeno 100% con gafas nasales a 3 litros por minuto. Tº 38.3 ºC.
Consciente, orientado y colaborador.
Auscultación cardíaca rítmico sin soplos. Auscultación pulmonar murmullo vesicular conservado.
Abdomen blando, depresible, con ruidos hidroaéreos conservados. No signos de irritación peritoneal.
Miembros inferiores no edemas, pulsos pedios presentes.
-Bioquímica: glucosa 250 mg/dl, urea 52 mg/dl, creatinina 1.5 mg/dl, Na 139 mEq/L, K 4.5 mEq/L, CPK 3623 UI/l, Troponina T Us 385 ng/L, GOT 53 UI/l, GPT 59 Ui/l.
-Radiología simple de tórax: sin hallazgos patológicos de interés.
-Electrocardiograma: ritmo sinusal a 117 lpm, imagen de bloqueo completo de rama derecha, con descenso de segmento ST de 2 m en V2-V5.
Evolución:
Ingresa con dolor que cede tras perfusión de nitroglicerina. Hipertenso (210/120 mmHg), taquicárdico a 150 latidos por minuto, sudoroso y con palidez mucocutánea. Se inicia levofloxacino como tratamiento antibiótico empírico por presentar tos con expectoración. Durante las primeras horas presentó hipertensión de difícil control.
En ecocardiografía realizada en la Unidad de Cuidados Intensivos presentó hipoquinesia anterior, con ventrículo izquierdo dilatado y fracción de eyección ligeramente deprimida. Se realizó coronariografía urgente a las 24 horas, con coronarias sin lesiones significativas. Seis horas tras el cateterismo, comenzó con nuevo episodio de dolor precordial intenso y emergencia hipertensiva, con cifras de tensión arterial de 300/150 mmHg. Se inició tratamiento con clevidipino en perfusión en dosis altas. Se asoció doxazosina con mala respuesta, alterando hipertensión con hipotensión. Sufrió deterioro respiratorio con edema agudo de pulmón y precisó intubación orotraqueal y conexión a ventilación mecánica.
Tras estabilización se solicitó Tomografía Computerizada (TC) abdominal sin contraste, donde se apreció una masa suprarrenal derecha de 5x4x4.5 cm, compatible con feocromocitoma dada la clínica del paciente (imágenes 1 y 2). Se inició bloqueo alfa con fenoxibenzamina y se solicitó estudio de catecolaminas en sangre y orina. Posteriormente se inicia tratamiento con calcioantagonistas por nuevos episodios de hipertensión arterial de difícil control.
Se completó estudio hormonal, con los siguientes resultados: cortisol 25, FSH 2, LH 2,5, PTH 32, Prolactina 25, TSH 4,9, Cromogranina A 266. Orina en 24 horas: metanefrinas totales 7429, normetafrinas 4649, vanilmandélico 14.3, Adrenalina 248 (N<18), noradrenalina 299 (N<76), dopamina 684.
Tras mejoría de cuadro de insuficiencia cardíaca con edema agudo de pulmón se inició tratamiento con propanolol intravenoso. Desde entonces, no presentó más episodios de hipertensión arterial con mejoría de la función renal hasta normalizarse. En ecocardiografía transtorácica posterior el VI no presentó dilatación, con fracción de eyección normal.
Finalmente es dado de alta al Servicio de Urología con interconsulta por parte de Endocrinología para el estudio y tratamiento de la masa suprarrenal. Posteriormente, se hizo gammagrafía con 123 I Metayodobencilguanidina (MIBG) observándose lesión nodular en glándula suprarrenal derecha con captación intensa del trazador, compatible feocromocitoma (imágenes 3 y 4). Tras tratamiento beta y alfa-bloqueante se intervino quirúrgicamente, realizándose adrenalectomía derecha, con buen resultado posterior.
Discusión:
El feocromocitoma es un tumor productor de catecolaminas que procede de las células cromafines del sistema nervioso simpático. Habitualmente deriva de la médula adrenal. Los feocromocitomas de localización extraadrenal se denominan paragangliomas y pueden originarse en cualquier lugar donde exista tejido cromafín (a lo largo de la cadena simpática ganglionar paraaórtica, en la pared de la vejiga urinaria y en la cadena ganglionar simpática en el cuello o mediastino)1.
Biosíntesis: La tirosina es transportada activamente a la médula adrenal, se hidroxila por la tirosina hidroxilasa, se acopla con tetrahidrobiopterina (cofactor), produciendo la L-DOPA; la descarboxilasa de L-aminoácido aromático produce dopamina, se almacena en los gránulos vesiculares y en la membrana, la dopamina b hidroxilasa con el ascorbato, cataliza la hidroxilación oxidativa produciendo norepinefrina que migran a la membrana celular y son secretadas parcialmente por exocitosis a la circulación. Después la norepinefrina es reciclada al citoplasma y la enzima feniletanolamina N-metiltransferasa y la S-adenosilmetionina (cofactor) metila el grupo amino, y sintetiza epinefrina. La norepinefrina y epinefrina pueden ser metabolizadas por catecol-ometiltransferasa y monoaminooxidasa produciendo normetanefrina y metanefrina.
Prevalencia: La incidencia es de 2-8 casos por un millón, es frecuente en la cuarta y quinta décadas de la vida. La prevalencia en la población hipertensa es de 0.3 a 1.9 %2,3 de las causas de hipertensión arterial secundaria.
Etiología: La frecuencia del feocromocitoma esporádico es 75% intraadrenal, 9-23% se desarrollan de tejido cromafin extraadrenal y se denominan paragangliomas4,5. La prevalencia de feocromocitoma maligno esporádico es 9%6. El 10% de feocromocitomas se presenta con metástasis al momento de su diagnóstico7.
El 25% de feocromocitoma esporádico sin patología puede ser portador de mutaciones en la línea germinal con tendencia a las patologías hereditarias: Neurofibromatosis tipo 1, enfermedad de Von Hippel- Lindau, neoplasia endocrina múltiple 2A, neoplasia endocrina múltiple 2B y feocromocitoma/paragangliomas
que establecen síndromes por mutaciones de genes en la subunidad D (SDHD), B (SDHB) y C (SDHC) de succinato deshidrogenada8,9,10
Neurofibromatosis tipo 1 (NF 1):
La transmisión es autosómica dominante. La frecuencia estimada es de 0.1-5.7%. El diagnóstico clínico de NF1 se establece en tener dos o más de siete criterios: Seis o más manchas de café con leche, dos o más neurofibromas cutáneos o neurofibromas plexiformes, neurofibromas inguinales, pecas en la región axilar, un glioma en el nervio óptico, familiares en primer grado con NF1.
La etiología consiste en mutación inactivante de neurofibromina, gen supresor de tumores, codifica la proteína activante GTPasa (en el cromosoma 17q11.211). De la neurofibromatosis tipo 1, el 90% son benignos (esporádicos 84%, bilaterales 10%, paragangliomas 6%). La mayoría son adultos y desarrollan hipertensión arterial.
Enfermedad Von Hippel –Lindau:
Es autosómica dominante, su incidencia es de 1 entre 3,600 nacidos vivos. VHL es producida por mutaciones en el gen del cromosoma 3p25-26. Una mutación en línea germinal desarrolla portadores de múltiples tumores12. El gen codifica para la proteína (pVHL) que suprime la formación de tumores y está implicada en la angiogénesis.
La expresión clínica de la enfermedad de VHL sigue cuatro subtipos con una función central en el feocromocitoma13,14,15. Los pacientes con VHL tipo 1 tienen pérdida de la función de pVHL, una mutación sin sentido y desarrollan hemangioblastoma en la retina, SNC y carcinoma renales, ellos no tienen riesgo de desarrollar feocromocitoma16,17. Los pacientes VHL tipo 2 tienen principalmente mutaciones sin sentido que desarrollan hemangioblastoma y feocromocitoma. Ellos tienen también un bajo riesgo (tipo 2A, tipo 2B alto riesgo para carcinoma de células renales). Un porcentaje de pacientes con VHL tipo 2C tienen feocromocitoma sin otro tumor.
Los portadores de VHL pueden presentarse como feocromocitomas esporádicos. En la patología VHL el feocromocitoma es la neoplasia más común (90%), aunque los paragangliomas han sido descritos, aproximadamente la mitad de los feocromocitomas son bilaterales18,19.
Neoplasia endocrina múltiple tipo 2:
Es autosómica dominante, se divide en 2A y 2B. MEN2A por una mutación inactivante de protooncogen RET en el cromosoma 10q11.2, implicada en la proliferación y apoptosis celular produciendo una activación constitutiva del receptor.
MEN2A: carcinoma medular de tiroides, hiperparatiroidismo primario y feocromocitoma.
MEN2B: feocromocitoma, carcinoma medular de tiroides y ganglioneuromas en las mucosas20. El feocromocitoma se presenta aproximadamente en 50% de los portadores y es localizado en la glándula adrenal. El feocromocitoma bilateral se desarrolla en el MEN2, su origen es frecuentemente asincrónico con un periodo subclínico hasta de 15 años21. Los feocromocitomas malignos son > 5% y generalmente grandes. El patrón de secreción de catecolaminas es principalmente por epinefrina, por lo tanto su fenotipo clínico está caracterizado por ansiedad, nerviosismo, palpitaciones, cefalea, más que el patrón común como manifestaciones cardiovasculares y hemodinámicas22.
Manifestaciones clínicas23, 24, 25, 26
Hipertensión arterial sistémica:
La hipertensión sostenida o paroxística es el signo clínico más común, lo presentan más de 90% de los pacientes. Los tumores que secretan norepinefrina se asocian con hipertensión sostenida. Los tumores que co-secretan norepinefrina y epinefrina están asociados a hipertensión episódica. Los tumores que producen exclusivamente epinefrina pueden producir más hipotensión que hipertensión.
La etiología de la hipertensión es la hipercatecolanemia en los receptores del sistema cardiovascular. Además existe un mecanismo en el SNC para el desarrollo de la hipertensión inducida por las catecolaminas: aumenta la concentración intraneuronal de catecolaminas almacenadas en vesículas, aumenta la frecuencia de los impulsos, y selectivamente desensibiliza receptores a2 adrenérgicos presinápticos, liberando cantidades excesivas de norepinefrina en el espacio sináptico y produce crisis hipertensiva.
Muchos feocromocitomas co-secretan norepinefrina y neuropéptido Y. El NPY tiene un potente efecto directo e indirecto sobre el sistema cardiovascular. En la circulación coronaria, independiente del sistema a adrenérgico. En algunos lechos vasculares el NPY no tiene un efecto vasoconstrictor pero potencia la vasoconstricción inducida por la norepinefrina. El NPY contribuye a hipertensión. En contraste pocos paragangliomas secretan NPY. Los feocromocitomas secretan muchos péptidos: PTHrP, ACTH, eritropoyetina, IL-6, la mayoría de los feocromocitomas secretan cromogranina A, la cual puede ser marcador tumoral27.
La tríada sintomática: Cefalea, diaforesis, y palpitaciones se encontró que tenía una sensibilidad de 90.9% y especificidad de 93.8%, lo cual a la luz de los conocimientos actuales no es cierto28. Sin embargo, el 8% pueden ser asintomáticos con formas familiares y grandes tumores quísticos. Aproximadamente 5% de los incidentalomas adrenales se han probado que son feocromocitomas funcionales. Las presentaciones agudas de feocromocitomas son espontáneas e inducidas por una cateterización vesical, anestesia y cirugía. La manifestación se produce por actividades benignas: esfuerzo, palpación abdominal, micción. A pesar de la hipertensión arterial, la cefalea, diaforesis y palpitaciones predominan como manifestaciones clínicas.
La administración de la terapia con b bloqueador no selectivo sin bloqueo a precedente puede precipitar una crisis con colapso hemodinámico.
Diagnóstico bioquímico:
Catecolaminas libres en la orina y metabolitos:
La determinación en catecolaminas libres y fraccionadas y sus metabolitos debe realizarse en orina de 24 h. Las concentraciones de norepinefrina > 170 mg, epinefrina > 35 mg y metanefrinas totales de 1.8 mg y ácido vainillilmandélico 11 mg en orina de 24 h establecen con mayor probabilidad el diagnóstico.
Catecolaminas plasmáticas:
La determinación de catecolaminas libres fraccionadas en el plasma en el feocromocitoma hereditario tiene sensibilidad de 69% y en el feocromocitoma esporádico de 92%, la especificidad es de 82 y 72%, respectivamente.
Metanefrinas plasmáticas:
En el HPLC la cuantía de normetraprefina y epinefrina en el caso de feocromocitoma hereditario, tiene sensibilidad de 97% y especificidad de 96% y en el esporádico la sensibilidad es de 99% y la especificidad de 82%, lo cual establece una gran superioridad de las metanefrinas libres en el plasma.
– Catecolaminas plasmáticas y urinarias:
Las catecolaminas son normalmente producidas por el sistema nervioso simpático, y la médula adrenal; no son específicos de feocromocitoma.
Los feocromocitomas secretan catecolaminas episódicamente; entre los episodios los niveles plasmáticos y urinarios son normales.
Los niveles plasmáticos y urinarios de catecolaminas son fisiológicos en 5 – 15% de pacientes con feocromocitomas.
Metanefrinas plasmáticas:
La cuantía de metanefrinas libres en plasma tiene ventajas debido a que su producción es constante e independiente de la exocitosis episódica de catecolaminas. Si las metanefrinas libres en plasma muestran concentraciones cuatro veces mayor que su límite superior, se confirma el diagnóstico de feocromocitoma. Pero si el valor de metanefrinas libres en el plasma se encuentra entre 3.9 veces y el límite superior normal, la siguiente etapa del diagnóstico es la prueba de supresión con clonidina.
Metanefrinas urinarias:
Si para la prueba principal, metanefrinas libres en plasma, no se tiene la técnica ni la experiencia en su realización, la determinación de metanefrinas fraccionadas en orina proporciona la segunda elección para el diagnóstico.
Hay fármacos que pueden alterar los resultados de catecolaminas y metanefrinas ( tabla 1).
Diagnóstico mediante pruebas de Imagen para la localización del feocromocitoma:
La tomografía axial computarizada (TAC) y la resonancia magnética (RM) son las principales pruebas de imagen para localización del feocromocitoma. Tiene una alta sensibilidad, pero una especificidad no óptima. La TAC y RM son los estudios de primera elección para la localización.
El feocromocitoma adrenal de 0.5 a 1 cm o más largos, o con metástasis de < 1 ó 2 cm pueden ser detectados por la TAC realizando cortes de 2 a 5 mm de grosor. Como la mayoría de feocromocitomas tienen un diámetro < 1 cm, son visualizados con la TAC. El coeficiente de atenuación sin contraste expresado en unidades Hounsfield (HU) está siendo utilizado para diferenciar los adenomas de otros tumores. Esto se basa en el hecho de que el contenido citoplasmático de lípidos es alto en los adenomas, pero escaso en los demás tumores29.
La sensibilidad de la TAC es de 85 a 94%, si el feocromocitoma está situado en la glándula adrenal30,31 . La sensibilidad para detectar lesiones extraadrenales, metastásicas y recurrentes es aproximadamente de 90%. Adicionalmente la especificidad de la TAC es limitada, se ha establecido que de aproximadamente 29 a 50%32,33,34. En los tumores adrenales la TAC con medio de contraste tiene una sensibilidad de 98% y una especificidad de 92%35,36 .
La RM debe ser sustituida por la TAC en niños, mujeres embarazadas y situaciones donde la exposición a radiación deba ser mínima.
Es especialmente útil, en el diagnóstico y seguimiento del feocromocitoma adrenal o la detección de metástasis con y sin gadolinio, que debe ser con secuencias T1. Así, el feocromocitoma emite señales semejantes al hígado, riñón y músculo y deben ser diferenciadas del tejido adiposo. La hipervascularidad del feocromocitoma tiene una apariencia brillante con secuencias T2. Particularmente, casi todos los feocromocitomas tienen una señal más intensa que el hígado o el músculo y frecuentemente son más intensas que la grasa. Entre las ventajas de la resonancia magnética está su alta sensibilidad para identificar feocromocitomas adrenales de 93 – 100%37, y otra ventaja es que expone al paciente a una baja radiación ionizante. La resonancia magnética es una buena modalidad de imagen para la detección de feocromocitomas intracardiacos, yuxtacardiacos, yuxtavasculares; el uso de secuencias T2 mejora la diferenciación del tejido adyacente al tumor.
La resonancia magnética se puede llevar a cabo con o sin el uso de agentes contrastados, por lo que no se tiene el riesgo de producir crisis hipertensiva. Comparado con la TAC parece dar una mejor valoración y mayor sensibilidad, especialmente si hay invasión vascular.
Imagen por ultrasonido:
Se considera que la especificidad para este estudio es de 60%. Es útil cuando el feocromocitoma es adrenal derecho e isodenso con el tejido hepático.
Estudios anatomofuncionales para la localización del feocromocitoma:
Después de la localización anatómica con la TAC y la RM, se procede a hacer los estudios funcionales que son [131I]-[123I] metayodobenzilguanidina, y el PET con varios ligandos39,40.
Imagen funcional con metayodobenzilguanidina:
La metayodobenzilguanidina es un arilguanidino que imita la norepinefrina. El marcaje radiactivo es realizado con los isótopos de yodo, 131I y 123I, en la posición meta del anillo benzoico. Semejante a la norepinefrina, la MIBG es captada por los tejidos simpatomedulares, principalmente el sistema de transporte noradrenérgico y dentro de las vesículas intracitoplasmáticas a través del sistema de transporte vesicular. El MIBG es así acumulado en el tejido adrenérgico. En la membrana plasmática y la captación vesicular es Na+ dependiente y puede ser influenciada por medicamentos tales como descongestionantes nasales, antihipertensivos, antidepresivos y cocaína; todos estos medicamentos deben retirarse tres días antes de realizar el estudio.
El estudio con [131I] MIBG tiene una sensibilidad de 77 – 90% y una alta especificidad de 95 a 100%41,42 . El isótopo de I, Br, C, F son marcadores de tejido. Los más usados han sido el 131I y el 123I.
Estudios de imagen funcional PET para la localización de feocromocitoma
La imagen de la tomografía por emisión de positrones (PET) es captada minutos u horas después de la inyección, es una sustancia de vida corta que emite positrones. La baja radiación de exposición y la resolución espacial, son las ventajas del PET, mientras que el costo y la disponibilidad limitada del radiofármaco y el equipo de PET, prohíbe ampliar su uso. En la valoración de los pacientes con feocromocitoma la [18F] FDG, [11C] hidroxiefedrina, ó [11C] efedrina han sido los más ampliamente usados43,44 . La [18F] DOPA es también ampliamente usada en NIH, [18F], lo mismo DA que se ha usado con éxito45,46
El aumento en el metabolismo de la glucosa, caracteriza a varios tejidos malignos, así, la captación de la glucosa marcada con [18F] tiene una vida media de 110 minutos, y en teoría, puede ser usada para la captación de imágenes de estos tumores.
Tratamiento farmacológico preoperatorio:
Fenoxibenzamina: es un bloqueador no competitivo y su unión es a receptores presinápticos a1 y a2. La compensación de los receptores ocupados es por la biosíntesis de nuevos receptores adrenérgicos. Los efectos colaterales son: hipotensión postural y taquicardia refleja. El efecto máximo se debe lograr durante 14 días, se inicia con 10 mg cada 12 h y el siguiente día, 15 mg cada 12 h y así hasta alcanzar el efecto terapéutico, la dosis máxima es de 1 mg/kg de peso, dividida en tres dosis.
Bloqueo a1 selectivo:La prazosina es un antagonista competitivo selectivo por los receptores a1, de corta duración de acción, por lo que su dosificación puede ser rápidamente ajustada, se inicia con 1 mg cada 8 h durante la primera semana, la segunda con 2 mg cada 8 h, la tercera con 3 mg cada 8 h, y si es necesario hasta 4 mg cada 8 h, la dosis máxima es de 12 mg al día.
Terazosina: antagonista competitivo selectivo de los receptores a1. Se inicia con 1 mg antes de acostarse, la dosis antihipertensiva es habitualmente de 1 a 5 mg cada día, con una dosis máxima de 20 mg/día.Doxazosina: es un inhibidor competitivo de los receptores a1, la dosis habitual es de 2 a 4 mg/día, se inicia con 1 mg/día durante una semana.
Según la respuesta, la dosis puede aumentarse a 2, 4, 8 y 16 mg al día como dosis máxima, con intervalos de una a dos semanas entre ellas.
Fentolamina:
Es un bloqueador de los receptores adrenérgicos a1 y a2 que pertenece al grupo de las b-imidazolinas. Es útil para el manejo transoperatorio. Se puede utilizar en bolo o en infusión intravenosa. El bolo de 5 a 10 mg se diluye en 10 mL de solución fisiológica al 0.9%. La infusión es de 300 mg por minuto. Los efectos colaterales son: hipotensión arterial, taquicardia refleja, espasmo cerebrovascular, estimulación del tracto gastrointestinal e hipoglucemia.
Bloqueo beta-adrenérgico:
El bloqueo b-adrenérgico controla las arritmias y la taquicardia. El bloqueo b-adrenérgico no selectivo con propranolol, se inicia con dosis de 40 mg cada 8 h, durante una semana y en la segunda semana 80 mg cada 8 h.
Los calcioantagonistas
Se utilizan con los bloqueadores a1, para controlar la hipertensión arterial sistémica. Para tratar las crisis es necesario: Nitroprusiato de sodio y fentolamina.
Sustitución de volumen plasmático, dos a tres litros de solución fisiológica a 0.9% un día antes de la cirugía.
Hemotransfusión aproximadamente de un litro o más, se realiza antes, durante y después de la cirugía. Una vez normotenso y sin inestabilidad hemodinámica, además del tratamiento simultáneo de comorbilidades se procede al tratamiento quirúrgico.
El abordaje quirúrgico depende de: La localización del feocromocitoma, y de la invasión vascular o capsular y su compromiso con las estructuras adyacentes, si se trata de un feocromocitoma bilateral, unilateral, genético, esporádico, así como del tamaño del feocromocitoma.
El tratamiento quirúrgico puede ser la adrenalectomía transperitoneal laparoscópica o adrenalectomía por cirugía abierta.
Diez días después de la cirugía se deben cuantificar las metanefrinas libres en el plasma y la catecolaminas urinarias, si éstas están normales, se continúa la vigilancia cada seis meses por dos años y después cada año indefinidamente. En caso de que exista hipermetanefrinemia se debe realizar búsqueda de metástasis con [123I] MIBG.
Conclusión:
El feocromocitoma es un tumor productor de catecolaminas que procede de las células cromafines del sistema nervioso simpático. Habitualmente deriva de la médula adrenal. Constituye una causa frecuente de incidentaloma suprarrenal, el 6.5% de dichos tumores. Es importnate sospechar, confirmar, localizar y resecar el feocromocitoma porque la hipertensión arterial asociada es curable, hay riesgo de muerte súbita, un 10% de los tumores hallados son malignos y la detección en los casos de afección familiar puede resultar en el diagnóstico precoz de otros miembros de la familia.